Descriere
Roswera 5 mg comprimate filmate
Roswera 10 mg comprimate filmate
Roswera 15 mg comprimate filmate
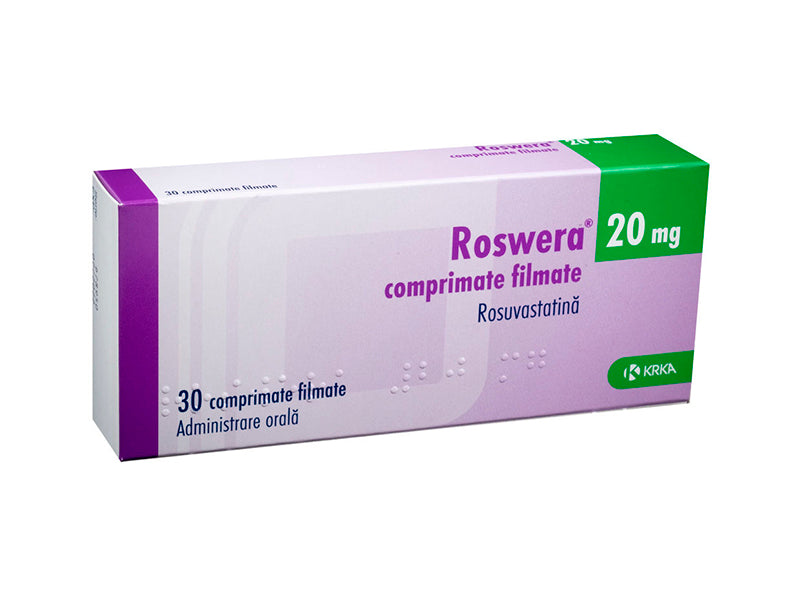
Roswera 20 mg comprimate filmate
Roswera 30 mg comprimate filmate
Roswera 40 mg comprimate filmate
COMPOZIŢIA CALITATIVĂ ŞI CANTITATIVĂ
Fiecare comprimat filmat conţine rosuvastatină 5 mg (sub formă de rosuvastatină calcică).
Fiecare comprimat filmat conţine 10 mg rosuvastatină (sub formă de rosuvastatină calcică).
Fiecare comprimat filmat conţine 15 mg rosuvastatină (sub formă de rosuvastatină calcică).
Fiecare comprimat filmat conţine 20 mg rosuvastatină (sub formă de rosuvastatină calcică).
Fiecare comprimat filmat conţine 30 mg rosuvastatină (sub formă de rosuvastatină calcică).
Fiecare comprimat filmat conţine 40 mg rosuvastatină (sub formă de rosuvastatină calcică).
Excipient(ţi) cu efect cunoscut:
Fiecare comprimat filmat de 5 mg conţine lactoză anhidră 41,9 mg.
Fiecare comprimat filmat de 10 mg conţine lactoză anhidră 41,9 mg.
Fiecare comprimat filmat de 15 mg conţine lactoză anhidră 62,9 mg.
Fiecare comprimat filmat de 20 mg conţine lactoză anhidră 83,8 mg.
Fiecare comprimat filmat de 30 mg conţine lactoză anhidră 125,7 mg.
Fiecare comprimat filmat de 40 mg conţine lactoză anhidră 167,6 mg.
FORMA FARMACEUTICĂ
Comprimat filmat.
5 mg: comprimate filmate de culoare albă, rotunde (diametrul de 7 mm), uşor biconvexe, cu margini teşite, marcate cu numărul 5 pe una dintre feţe.
10 mg: comprimate filmate de culoare albă, rotunde (diametrul de 7,5 mm), uşor biconvexe, cu margini teşite, marcate cu numărul 10 pe una dintre feţe.
15 mg: comprimate filmate de culoare albă, rotunde (diametrul de 9 mm), uşor biconvexe, cu margini teşite, marcate cu numărul 15 pe una dintre feţe.
20 mg: comprimate filmate de culoare albă, rotunde (diametrul de 10 mm), cu margini teşite.
30 mg: comprimate filmate de culoare albă, biconvexe, alungite, marcate pe ambele feţe (dimensiuni:15 mm x 8 mm). Linia mediană are numai rolul de a uşura ruperea comprimatului pentru a fi înghiţit uşor şi nu de divizare în doze egale.
40 mg: comprimate filmate de culoare albă, biconvexe, alungite (dimensiuni: 16 mm x 8,5 mm).
DATE CLINICE
Indicaţii terapeutice
Tratamentul hipercolesterolemiei
Adulţi, adolescenţi şi copii cu vârsta de 10 ani sau peste, cu hipercolesterolemie primară (tip IIa, inclusiv hipercolesterolemia familială - forma heterozigotă) sau dislipidemie mixtă (tip IIb), ca adjuvant al dietei la pacienţii care nu răspund adecvat numai la dietă şi la alte metode de tratament non-farmacologic (cum sunt exerciţiile fizice, scăderea în greutate).
Hipercolesterolemie familială - forma homozigotă, ca tratament adjuvant al dietei şi al altor metode de scădere a lipidemiei (de exemplu afereza LDL) sau în cazurile în care aceste tratamente nu sunt adecvate.
Prevenţia evenimentelor cardiovasculare Prevenţia evenimentelor cardiovasculare majore la pacienţii cu risc major pentru un prim eveniment cardiovascular (vezi pct. 5.1), ca tratament adjuvant în controlul altor factori de risc.
Doze şi mod de administrare
Doze
Înainte de începerea tratamentului, pacientul trebuie să urmeze un regim hipocolesterolemiant standard care trebuie continuat şi în timpul tratamentului. Doza trebuie adaptată pentru fiecare pacient, în funcţie de obiectivul tratamentului şi de răspunsul pacientului, respectând recomandările din ghidurile actuale de tratament.
Roswera poate fi administrat în orice moment al zilei, cu sau fără alimente.
Este posibil să nu fie disponibile toate concentraţiile de Roswera.
Tratamentul hipercolesterolemiei
Doza iniţială recomandată este de 5 sau 10 mg administrată oral, o dată pe zi, atât la pacienţii care iau pentru prima dată statine, cât şi la pacienţii care trec de pe un tratament cu alt inhibitor de HMG CoA reductază. Alegerea dozei iniţiale se face în funcţie de nivelul individual al colesterolemiei şi aprecierea riscului cardiovascular, precum şi de riscul potenţial al reacţiilor adverse. Dacă este necesar, doza poate fi crescută după 4 săptămâni la 20 mg.
Datorită creşterii incidenţei reacţiilor adverse la utilizarea dozei de 40 mg, comparativ cu dozele mai scăzute, creşterea dozei de la 20 mg la 30 mg sau până la doza maximă de 40 mg trebuie luată în considerare numai la pacienţii cu hipercolesterolemie severă şi risc cardiovascular crescut (în particular la pacienţii cu hipercolesterolemie familială), care nu ating valoarea stabilită ca obiectiv al tratamentului cu doza de 20 mg, aceştia urmând a fi monitorizaţi prin teste periodice.
La iniţierea tratamentului cu dozele de 30 mg saur 40 mg este necesară monitorizare de specialitate.
Prevenţia evenimentelor cardiovasculare
În studiul clinic al reducerii riscului de evenimente cardiovasculare, doza utilizată a fost de 20 mg pe zi.
Pacienţi vârstnici
La pacienţii cu vârsta >70 ani se recomandă o doză iniţială de 5 mg.
Pacienţi cu insuficienţă renală
Nu este necesară ajustarea dozelor la pacienţi cu insuficienţă renală uşoară sau moderată. Doza iniţială recomandată la pacienţi cu insuficienţă renala moderată (clearance-ul creatininei <60 ml/min) este de 5 mg. Utilizarea dozelor de 30 mg şi 40 mg este contraindicată la pacienţii cu insuficienţă renala moderată. Utilizarea Roswera la pacienţi cu insuficienţă renală severă este contraindicată la orice doză.
Pacienţi cu insuficienţă hepatică
Pentru pacienţii cu scoruri Child-Pugh de 7 sau mai mici nu s-a constatat creşterea expunerii sistemice la rosuvastatină. Totuşi, creşterea expunerii sistemice a fost observată la pacienţi cu scoruri ChildPugh de 8 şi 9. La aceşti pacienţi trebuie avută în vedere evaluarea funcţiei renale. Nu există experienţă clinică la pacienţi cu scoruri Child-Pugh peste 9. Roswera este contraindicat la pacienţii cu boală hepatică activă.
Copii şi adolescenţi
Utilizarea la copii trebuie recomandată de către medicii de specialitate.
Copii şi adolescenţi cu vârsta de 10 până la 17 ani (băieţi: stadiul Tanner II sau peste, iar fetele la cel puţin 1 an după menarhă)
La copii şi adolescenţi cu hipercolesterolemie familială heterozigotă, doza uzuală iniţială este de 5 mg pe zi. Intervalul de dozare uzual este 520 mg oral, o dată pe zi.
Creşterea dozelor trebuie efectuată în funcţie de răspunsul la tratament şi tolerabilitatea individuală a copiilor, conform recomandărilor ghidurilor de tratament pediatric. Copiii și adolescenții trebuie să efectueze tratament dietetic hipocolesterolemiant standard înainte de inițierea tratamentului cu rosuvastatină; regimul alimentar trebuie continuat pe toată durata tratamentului cu rosuvastatină.
Siguranţa şi eficacitatea tratamentului cu doze mai mari de 20 mg nu au fost încă stabilite la aceste grupe de populaţie. Comprimatele de 30 şi 40 mg nu sunt recomandate la copii.
Copii cu vârsta sub 10 ani
Experienţa clinică este limitată la un număr mic de copii (cu vârste de 8 ani şi până la 10 ani) cu hipercolesterolemie familială - forma homozigotă. De aceea, Roswera nu este recomandat la copii cu vârsta sub 10 ani.
Rasă
La subiecţii asiatici a fost observată creşterea expunerii sistemice. La pacienţii de origine asiatică, se recomandă o doză iniţială de 5 mg. Dozele de 30 şi 40 mg sunt contraindicate la aceşti pacienţi.
Polimorfism genetic
Se ştie că prezenţa anumitor tipuri de polimorfism genetic poate duce la creşterea concentraţiilor plasmatice ale rosuvastatinei (vezi pct. 5.2). La pacienţii cu astfel de tipuri cunoscute de polimorfism, se recomandă administrarea unor doze mai mici de rosuvastatină.
Doze la pacienţi cu factori de risc de miopatie
La pacienţi cu factori de risc de miopatie doza inţială recomandată este de 5 mg. Dozele de 30 şi 40 mg sunt contraindicate la unii dintre aceşti pacienţi.
Tratament concomitent
Rosuvastatina reprezintă substrat al unor proteine de transport (de exemplu, OATP1B1 şi BCRP). Riscul de miopatie (incluzând rabdomioliză) este crescut în cazul administrării concomitente a rosuvastatinei cu anumite medicamente ce pot creşte concentraţiile plasmatice ale rosuvastatinei, din cauza interacţiunilor cu aceste proteine transportoare (de exemplu, ciclosporina şi anumiţi inhibitopri ai proteazelor, inclusiv combinaţi de ritonavir cu atazanavir, lopinavir şi/sau tipranavir. Atunci când este posibil, se recomandă luarea în consideraţie a unui tratament alternativ şi, dacă este necesar, terapia cu rosuvastatină poate fi întreruptă. În situaţiile în care administrarea concomitentă a acestor medicamnte şi rosuvastatină nu poate fi evitată, trebuie evaluat raportul beneficiu/risc al tratamentului concomitent şi ajustarea cu atenţie a dozelor de rosuvastatin.
Contraindicaţii
Roswera este contraindicată:
- la pacienţii cu hipersensibilitate la substanţa activă sau la oricare dintre excipienţii enumeraţi
- la pacienţi cu boală hepatică activă, incluzând creşterea inexplicabilă, persistentă, a transaminazelor plasmatice şi orice creştere a transaminazelor plasmatice care depăşeşte de 3 ori limita superioară a normalului (ULN)
- la pacienţi cu insuficienţă renală severă (clearance al creatininei <30 ml/min).
- la pacienţi cu miopatie
- la pacienţi care primesc tratament concomitent cu ciclosporină
- în timpul sarcinii şi alăptării şi la femei aflate în perioada fertilă care nu utilizează măsuri contraceptive corespunzătoare.
Dozele de 30 şi 40 mg sunt contraindicate la pacienţi cu factori de risc de miopatie/rabdomioliză.
Astfel de factori de risc sunt:
- insuficienţa renală moderată (clearance-ul creatininei <60 ml/min)
- hipotiroidie
- antecedente personale sau familiale de afecţiuni ereditare musculare
- istoric de toxicitate musculară la un alt inhibitor al HMG-CoA reductază sau fibraţi
- consum exagerat de alcool etilic
- situaţii în care nivelele plasmatice ale medicamentului pot creşte
- pacienţi cu origine asiatică - utilizare concomitentă de fibraţi.
Atenţionări şi precauţii speciale pentru utilizare
Efecte renale
Proteinuria, evidenţiată prin teste de tip "dipstick" şi de cele mai multe ori de origine tubulară, a fost observată la pacienţii trataţi cu doze mai mari de rosuvastatină în particular 40 mg, şi în cele mai multe cazuri a fost tranzitorie sau intermitentă. Proteinuria nu s-a demonstrat a fi un factor predictiv al unei boli renale acute sau progresive. Incidenţa raportărilor de reacţii adverse severe renale la utilizarea dozei de 40 mg, în perioada de după punerea pe piaţă a medicamentului, este mai mare. În cazul pacienţilor trataţi cu doze de 30 mg sau 40 mg, la efectuarea controlului de rutină trebuie avută în vedere şi evaluarea funcţiei renale.
Efecte la nivelul musculaturii scheletice
La pacienţii trataţi cu rosuvastatină au fost raportate efecte asupra muşchilor scheletici, ca mialgie, miopatie şi, rareori, rabdomioliză la toate dozele şi în special la doze de peste 20 mg. În foarte rare cazuri, a fost raportată rabdomioliza la asocierea de ezetimib şi inhibitori ai HMG-CoA reductazei. Interacţiunea farmacodinamică nu poate fi exclusă şi la utilizarea acestei asocieri, este necesară precauţie.
Ca şi în cazul altor inhibitori ai HMG-CoA reductazei, incidenţa rabdomiolizei asociate cu utilizarea Rowsera este mai mare la utilizarea dozei de 40 mg, în perioada de după punerea pe piaţă a medicamentului.
Determinarea valorilor creatinkinazei
Creatinkinaza (CK) nu trebuie măsurată după efectuarea unor exerciţii fizice intense sau în prezenţa unei cauze evidente care ar putea să ducă la creşterea valorilor CK şi care ar putea modifica interpretarea rezultatului. În cazul în care concentraţiile plasmatice iniţiale ale CK sunt crescute în mod semnificativ (>5 x ULN), trebuie efectuat un test de confirmare în următoarele 5-7 zile. Dacă repetarea testului confirmă o valoare iniţială de >5 x ULN, atunci tratamentul nu trebuie început.
Înainte de tratament
Similar altor inhibitori ai HMG-CoA reductazei, Roswera trebuie recomandat cu prudenţă la pacienţii care prezintă factori predispozanţi pentru miopatie/rabdomioliză:
- insuficienţă renală
- hipotiroidie
- antecedente personale sau heredocolaterale de afecţiuni musculare ereditare
- antecedente de toxicitate musculară la administrarea altor inhibitori ai HMG-CoA reductazei sau a fibraţilor
- consum exagerat de alcool etilic
- vârstă >70 ani - situaţii în care poate apare o creştere a concentraţiilor plasmatice
- utilizarea concomitentă a fibraţilor.
La asemenea pacienţi, riscul trebuie evaluat în funcţie de posibilele beneficii ale tratamentului şi este recomandată monitorizarea clinică. Dacă nivelele CK sunt semnificativ crescute faţă de valorile bazale (>5 x ULN) tratamentul nu trebuie iniţiat.
În timpul tratamentului
Pacienţilor trebuie să li se recomande să raporteze imediat durerile musculare inexplicabile, hipotonia musculară sau crampele, mai ales dacă acestea se asociază cu stare de rău general sau febră. La aceşti pacienţi trebuie măsurate concentraţiile plasmatice ale CK. Tratamentul trebuie întrerupt dacă: concentraţiile plasmatice ale CK sunt mult crescute (>5 x ULN) sau dacă simptomele musculare sunt severe şi determină un disconfort zilnic (chiar şi în cazul în care concentraţiile plasmatice ale CK sunt ≤5 ori limita superioară a normalului). Dacă simptomele se remit şi concentraţiile plasmatice ale CK revin la normal, atunci trebuie avută în vedere reluarea tratamentului cu Roswera sau cu un alt inhibitor de HMG-CoA reductază, în cea mai mică doză şi sub o monitorizare atentă. Monitorizarea de rutină a concentraţiilor plasmatice ale CK la pacienţii asimptomatici nu este justificată. Au fost raportate cazuri foarte rare de miopatie necrozantă prin mecanism imunitar (IMNM) în timpul sau după tratamentul cu statine, incusiv rosuvastatină. IMNM este caracterizat clinic prin astenie musculară proximală şi creşterea concentraţiilor plasmatice ale CK, care persistă, chiar dacă tratamentul cu statine este întrerupt.
În studiile clinice, la numărul mic de pacienţi trataţi cu rosuvastatină şi un alt tratament concomitent, nu s-a înregistrat o incidenţă mai mare a efectelor asupra musculaturii scheletice. Cu toate acestea, la pacienţii trataţi cu alţi inhibitori ai HMG-CoA reductazei, concomitent cu fibraţi cum este gemfibrozilul, ciclosporină, acid nicotinic, antifungice azolice, inhibitori ai proteazelor şi antibiotice macrolidice s-a constatat o incidenţă crescută a miozitei şi miopatiei. Gemfibrozilul creşte riscul de miopatie atunci când este administrat concomitent cu unii inhibitori ai HMG-CoA reductazei. De aceea, asocierea Roswera şi gemfibrozil nu este recomandată. Beneficiul privind scăderea suplimentară a concentraţiilor plasmatice de lipide prin administrarea asociată de Roswera şi fibraţi sau niacină trebuie atent evaluat faţă de potenţialele riscuri ale acestei asocieri. Dozele de 30 mg şi 40 mg sunt contraindicate la utilizare concomitentă cu fibraţi.
Roswera nu trebuie utilizat la nici un pacient care prezintă o afecţiune acută, severă, sugestivă pentru miopatie sau cu predispoziţie de a dezvolta insuficienţă renală secundară rabdomiolizei (de exemplu sepsis, hipotensiune arterială, intervenţie chirurgicală majoră, traumatisme, tulburări severe metabolice, endocrine şi electrolitice; crize convulsive necontrolate).
Efecte hepatice
Ca şi în cazul altor inhibitori ai HMG-CoA reductazei, Roswera trebuie utilizat cu prudenţă la pacienţii care consumă cantităţi excesive de alcool etilic şi/sau au antecedente de boală hepatică.
Se recomandă efectuarea de teste funcţionale hepatice înainte de începerea tratamentului şi la 3 luni după începerea tratamentului. În cazul în care concentraţia plasmatică a transaminazelor este de 3 ori mai mare decât limita superioară a normalului, tratamentul cu Roswera trebuie întrerupt sau doza trebuie redusă. Incidenţa reacţiilor hepatice severe (în principal constând în creşterea transaminazelor hepatice) este mai mare în perioada de după punerea pe piaţă a medicamentului, pentru doza de 40 mg.
La pacienţii cu hipercolesterolemie secundară determinate de hipotiroidism sau sindrom nefrotic, afecţiunea de bază trebuie tratată înaintea începerii tratamentului cu Roswera.
Rasa Rezultatele studiilor de farmacocinetică arată o creştere a expunerii la pacienţii de rasă asiatică, comparativ cu cei de rasă albă.
Inhibitorii de proteaze
La pacienţii la care s-au administrat concomitent rosuvastatină şi diverşi inhibitori ai proteazelor în asociere cu ritonavir, a fost observată creşterea concentraţiilor plasmatice ale rosuvastatinei. La iniţierea şi creşterea dozelor de rosuvastatină la pacienţii cu HIV trataţi concomitent cu inhibitori ai proteazelor, trebuie luat în considerare atât beneficiul scăderii lipidemiei de către rosuvastatină, cât şi creşterea potenţială a concentraţiilor plasmatice ale rosuvastatinei. Utilizarea concomitentă a rosuvastatinei şi inhibitorilor proteazelor nu este recomandată, decât cu.Utilizarea concomitentă cu inhibitorii de proteaze se poate face numai cu scăderea dozelor de rosuvastatină.
Pneumonie interstiţială
La utilizarea unor statine, în special la utilizare îndelungată, au fost raportate cazuri excepţional de rare de pneumonie interstiţială (vezi pct. 4.8). Tabloul clinic poate include dispnee, tuse neproductivă şi deteriorarea stării generale (fatigabilitate, pierdere în greutate şi hipertermie). Dacă se suspectează că un pacient dezvoltă o pneumonie interstiţială, tratamentul cu statine trebuie întrerupt.
Diabet zaharat
Există dovezi care sugerează că statinele cresc glicemia şi, la unii pacienţi cu risc crescut de apariţie a diabetului zaharat, pot produce hiperglicemie cu valori care să necesite măsuri considerate de rutină la pacienţii cu diabet zaharat diagnosticat. Totuşi, riscul de apariţie a diabetului zaharat este depăşit de beneficiul reducerii riscului cardiovascular şi, prin urmare, nu există un motiv pentru întreruperea tratamentului cu statine. Pacienţii cu risc crescut (valori ale glicemiei în condiţii de repus alimentar între 5,6 şi 6,9 mmol/l, IMC>30 kg/m2, valori crescute ale trigliceridemiei, hipertensiune arterială) trebuie monitorizaţi clinic şi paraclinic în acord cu ghidurile naţionale. În studiul clinic JUPITER, frecvenţa globală raportată de apariţie a diabetului zaharat a fost de 2,8% la rosuvastatină şi de 2,3% la placebo, mai ales la pacienţii cu valori ale glicemiei între 5,6 şi 6,9 mmol/l în condiţii de repaus alimentar.
Copii şi adolescenţi
Evaluarea înalţimii, greutăţii corporale, indexului masei corporale şi a caracteristicilor sexuale secundare conform scalei Tanner la pacienţii copii cu vârsta de 10 până la 17 ani în tratament cu rosuvastatină este limitată la perioada de 1 an. După un studiu de 52 săptămâni, nu au fost observate efecte asupra creşterii, greutăţii corporale, IMC sau maturizării sexuale. Datele clinice la copii şi adolescenţi sunt limitate, iar efectele rosuvastatinei pe perioade îndelungate (>1 an) asupra pubertăţii sunt necunoscute. Într-un studiu clinic la copii şi adolescenţi în tratament cu rosuvastatină timp de 52 săptămâni, creşterile CK peste ULN x 10 şi simptomele musculare apărute după exerciţii fizice sau activitate fizică crescută, au apărut mai frecvent, comparativ cu studiile clinice la adulţi.
Roswera comprimate filmate conţine lactoză. Pacienţii cu afecţiuni ereditare rare de intoleranţă la galactoză, deficit de lactază (Lapp) sau sindrom de malabsorbţie la glucoză-galactoză nu trebuie să utilizeze acest medicament.
Interacţiuni cu alte medicamente şi alte forme de interacţiune
Efectele altor medicamente asupra rosuvastatinei
Inhibitorii proteinelor de transport
Rosuvastatina este substrat al anumitor proteine de transport, incuzând transportorul hepatic de captare OATP1B1 şi a transportorului hepatic de eflux BCRP. Administrarea concomitentă a rosuvastatinei împreună cu medicamente care inhibă activitatea proteinelor de transport poate duce la creşterea concentraţiilor plasmatice ale rosuvastatinei şi la creşterea riscului de miopatie.
Ciclosporină
În timpul tratamentului concomitent cu rosuvastatină şi ciclosporină, valorile ariei de sub curba concentraţiei plasmatice în funcţie de timp (ASC) ale rosuvastatinei au fost în medie de 7 ori mai mari decât cele observate la voluntarii sănătoşi (vezi tabelul 1). Rosuvastatina este contraindicată la pacienţii la care se administrează concomitent ciclosporină. Administrarea concomitentă nu a afectat concentraţiile plasmatice ale ciclosporinei.
Inhibitorii proteazelor
Cu toate că nu se cunoaşte exact mecanismul interacţiunii, utilizarea concomitentă a unui inhibitor de protează poate determina o creştere puternică a concentraţiilor plasmatice ale rosuvastatinei. De exemplu, într-un studiu de farmacocinetică, administrarea concomitentă la voluntari tineri de rosuvastatină 10 mg şi o combinaţie de doi inhibitori ai proteazelor (atazanavir 300 mg şi ritonavir100 mg), a fost asociată cu o creştere de aproximativ trei ori, respectiv şapte ori, a ASC şi, respectiv, Cmax a rosuvastatinei la starea de echilibru. Utilizarea concomitentă de rosuvastatină şi unele combinaţii de inhibitori ai proteazelor poate fi luată în considerare numai după ajustarea atentă a dozelor de rosuvastatină, în funcţie de creşterea aşteptată a concentraţiilor plasmatice ale rosuvastatinei.
Gemfibrozil şi alte hipolipemiante
Utilizarea concomitentă de rosuvastatină şi gemfibrozil a determinat creşterea de 2 ori a Cmax şi ASC pentru rosuvastatină. Pe baza datelor obţinute din studiile de interacţiune specifică nu sunt de aşteptat interacţiuni clinic semnificative în cazul administrării concomitente de fenofibrat, totuşi poate apărea o interacţiune farmacodinamică. Gemfibrozilul, fenofibratul, alţi fibraţi şi doze hipolipemiante (≥ 1g/zi) de niacină (acid nicotinic) cresc riscul de miopatie atunci când sunt administrate concomitent cu inhibitori ai HMG-CoA reductazei, probabil datorită faptului că aceştia pot produce miopatie şi în monoterapie. Dozele de 30 mg şi 40 mg sunt contraindicate la utilizarea concomitentă a fibraţilor. De asemenea, aceşti pacienţi trebuie să utilizeze doza iniţială de 5 mg. Ezetimib
Utilizarea concomitentă de rosuvastatină 10 mg şi ezetimib 10 mg a determinat o creştere de 1,2 ori a ASC pentru rosuvastatină la pacienţii cu hipercolesterolemie. Totuşi, nu poate fi exclusă o interacţiune farmacodinamică, privitor la reacţiile adverse, între Roswera şi ezetimib.
Antiacide
Administrarea simultană de rosuvastatină şi o suspensie conţinând un antiacid cu hidroxid de aluminiu şi magneziu a determinat scăderea concentraţiei plasmatice a rosuvastatinei de aproximativ 50%. Acest efect a fost mai mic atunci când antiacidul a fost administrat la 2 ore după Rowsera. Nu a fost studiată importanţa clinică a acestei interacţiuni.
Eritromicină
Administrarea simultană de rosuvastatină şi eritromicină a dus la scăderea cu 20% a ASC şi scăderea cu 30% a Cmax a rosuvastatinei. Această interacţiune poate fi determinată de creşterea motilităţii intestinale de către eritromicină.
Enzimele citocromului P450
Rezultatele din studiile in vitro şi in vivo arată că rosuvastatina nu este nici inhibitor şi nici inductor al izoenzimelor citocromului P450. În plus, rosuvastatina este un substrat slab pentru aceste enzime. De aceea, nu sunt aşteptate interacţiuni medicamentoase depinzând de metabolizarea mediată de citocromul P450. Nu s-au observat interacţiuni clinic semnificative nici între rosuvastatină şi fluconazol (un inhibitor al CYP2C9 şi CYP3A4) sau rosuvastatină şi ketoconazol (un inhibitor al CYP2A6 şi CYP3A4).
Interacţiuni care necesită ajustarea dozei de rosuvastatină
Atunci când este necesară administrarea concomitentă de rosuvastatină şi alte medicamente care determină creşterea cunoscută a concentraţiilor plasmatice ale rosuvastatinei, dozele de rosuvastatină trebuie scăzute. În cazul în care creşterea aşteptată a ASC este de aproximativ 2 ori sau mai mare, tratamentul trebuie iniţiat cu doza zilnică unică de 5 mg rosuvastatină. Doza zilnică maximă de rosuvastatină trebuie ajustată astfel încât concentraţiile plasmatice de rosuvastatină aşteptate să nu depăşească pe cele care apar după administrarea de rosuvastatină 40 mg pe zi, în monoterapie; de exemplu, la administrarea concomitentă cu gemfibrozil, trebuie administrată o doză zilnică de 20 mg rosuvastatină (creştere de 1,9 ori a ASC) şi de 10 mg rosuvastatină, în cazul administrării concomitente cu asocierea atazanavir/ritonavir (creştere de 3,1 ori a ASC).
Efectul rosuvastatinei asupra unor medicamente administrare concomitent
Antagonişti de vitamină K
Ca şi în cazul altor inhibitori ai HMG-CoA reductazei, iniţierea tratamentului sau creşterea dozei de Roswera la pacienţi trataţi concomitent cu antagonişti de vitamină K (de exemplu warfarină sau alt anticoagulant cumarinic) pot duce la creşterea International Normalised Ratio (INR). Întreruperea tratamentului sau scăderea dozelor de Roswera poate duce la scăderea INR-ului. În astfel de situaţii, se impune monitorizarea valorilor INR.
Contraceptive orale/tratament de substituţie hormonală (HRT-hormone replacement therapy)
Administrarea concomitentă de rosuvastatină şi un contraceptiv oral a determinat creşterea ASC pentru etinilestradiol şi norgestrel cu 26% şi, respectiv, 34%. Această creştere a concentraţiilor plasmatice trebuie avută în vedere când se aleg dozele de contraceptive orale. Nu sunt disponibile date de farmacocinetică pentru subiecţii care utilizează concomitent rosuvastatină şi HRT şi, de aceea, un efect similar nu poate fi exclus. Cu toate acestea, în studiile clinice, asocierea a fost frecvent utilizată la femei şi a fost bine tolerată.
Alte produse medicamentoase
Pe baza datelor obţinute din studiile de interacţiune specifică, nu sunt de aşteptat interacţiuni clinic semnificative în cazul administrării concomitente de digoxină.
Copii şi adolescenţi
Au fost efectuate studii de interacţiune medicamentoasă numai la adulţi. Nivelul interacţiunilor medicamentoase la copii şi adolescenţi nu este cunoscut.
Fertilitatea, sarcina şi alăptarea
Roswera este contraindicat în sarcină şi alăptare.
Sarcina
Femeile cu potenţial fertil trebuie să utilizeze metode contraceptive corespunzătoare.
Deoarece colesterolul şi alţi produşi ai biosintezei colesterolului sunt esenţiali pentru dezvoltarea fătului, riscul potenţial al inhibării HMG-CoA reductazei depăşeşte avantajul tratamentului în timpul sarcinii. Studiile efectuate la animale aduc informaţii limitate despre toxicitatea asupra funcţiei de reproducere. Dacă o pacientă rămâne însărcinată în timpul utilizării acestui produs, tratamentului trebuie întrerupt imediat.
Alăptarea
Rosuvastatina se excretă în lapte, la şobolan. Nu există date privind excreţia în lapte, la om.
Efecte asupra capacităţii de a conduce vehicule şi de a folosi utilaje
Nu s-au efectuat studii care să determine efectul administrării rosuvastatinei asupra capacităţii de a conduce vehicule sau de a folosi utilaje. Cu toate acestea, pe baza proprietăţilor sale farmacodinamice, este puţin probabil ca rosuvastatina să afecteze aceste abilităţi. La conducerea vehiculelor sau la folosirea utilajelor, trebuie avut în vedere că în timpul tratamentului pot să apară ameţeli.
Reacţii adverse
Sumar al profilului de siguranţă
Reacţiile adverse care apar la administrarea rosuvastatinei sunt, în general, uşoare şi tranzitorii. În studiile clinice controlate, mai puţin de 4% dintre pacienţii trataţi cu rosuvastatină au fost excluşi din cauza reacţiilor adverse.
Lista tabelară a reacţiilor adverse
Pe baza datelor din studiile clinice şi experienţa vastă după punerea pe piaţă, următorul tabel prezintă profilul reacţiilor adverse pentru rosuvastatină. Reacţiile adverse enumerate mai jos sunt clasificate în funcţie de frecvenţă şi sisteme de organe (SO).
- Foarte frecvente (>1/10)
- Frecvente (>1/100 la <1/10)
- Mai puţin frecvente (>1/1000 la <1/100)
- Rare (>1/10000 la <1/1000)
- Foarte rare (<1/10000),
- Cu frecvenţă necunoscută (nu poate fi estimată din datele disponibile)
Reacţiile adverse pe baza datelor din studiile clinice
-Tulburări hematologice şi limfatice
-Trombocitopenie
-Tulburări ale sistemului imunitar
-Reacţii de hipersenbilitate, inclusiv angioedem
-Tulburări endocrine
-Diabet zaharat1
-Tulburări psihice
-Depresie
-Tulburări ale sistemului nervos
-Cefalee Ameţeli
-Polineuropatie Pierderea memoriei
-Neuropatie periferică Tulburări ale somnului (inclusiv insomnie şi coşmaruri)
-Tulburări respiratorii, toracice şi mediastinale
-Tuse Dispnee
-Tulburări gastrointestinale
-Constipaţie,Greaţă,Durere abdominală
-Pancreatită,Diaree
-Tulburări hepatobiliare
-Creşterea concentraţiilor transaminazelor hepatice
-Icter Hepatită
-Afecţiuni cutanate şi ale ţesutului subcutanat
-Prurit Erupţie cutanată tranzitorie Urticarie
- StevensJohnson syndrome
-Tulburări musculoscheletice şi ale ţesutului conjunctiv
-Mialgie Miopatie (inclusiv miozită) Rabdomioliză
-Artralgie Miopatie necrozantă mediată imunitar
-Tulburări tendinoase, uneori complicate prin ruptură de tendon
-Tulburări renale şi ale căilor urinare
-Hematurie
-Tulburări ale aparatului genital şi sânului
-Ginecomastie
-Tulburări generale şi la nivelul locului de administrare
-Astenie
-Edeme
Copii şi adolescenţi
Creşterea creatinkinazei de peste 10 x ULN şi simptomele musculare după efort fizic sau creşterea activităţii fizice au fost observate mai frecvent într-un studiu clinic de 52 săptămâni, în care s-au facut comparaţii între copii şi adulţi (vezi pct. 4.4). Din alt punct d evedere, profilul siguranţei rosuvastatinei a fost similar la copii şi adolescenţi, comparativ cu adulţii.
Raportarea reacţiilor adverse suspectate
Raportarea reacţiilor adverse suspectate după autorizarea medicamentului este importantă.
Supradozaj
Nu există tratament specific în caz de supradozaj. În caz de supradozaj, pacientul trebuie tratat simptomatic şi trebuie instituite măsurile de susţinere adecvate. Se recomandă monitorizarea funcţiei hepatice şi a concentraţiilor plasmatice de CK. Este puţin probabil ca hemodializa să aducă vreun beneficiu.
PROPRIETĂŢI FARMACOLOGICE
Proprietăţi farmacodinamice
Grupa farmacoterapeutică: hipocolesterolemiante şi hipotrigliceridemiante; inhibitori ai HMG-CoA reductazei, codul ATC: C10AA07.
Mecanism de acţiune
Rosuvastatina este un inhibitor selectiv şi competitiv al HMG-CoA reductazei, enzima cheie în procesul de transformare a 3-hidroxil-3-metilglutaril coenzima A la mevalonat, un precursor al colesterolului. Locul principal de acţiune al rosuvastatinei este ficatul, organul ţintă pentru scăderea olesterolului.
Rosuvastatina creşte numărul receptorilor LDL de pe suprafaţa celulelor hepatice, crescând captarea şi catabolismul LDL şi inhibă sinteza hepatică de VLDL, reducând în acest mod numărul total al particulelor de VLDL şi LDL
Efecte farmacodinamice
Rosuvastatina scade concentraţiilor crescute de LDL-colesterol, colesterol total şi trigliceride şi creşte HDL-colesterolul. De asemenea, rosuvastatina scade Apo-B, non HDL-C, VLDL-C, VLDL-TG, rapoartele LDL-C/HDL-C, C total/HDL-C, non HDL-C/HDL-C, Apo-B/ApoA-I.
Efectul terapeutic se obţine într-o săptămână de la începerea tratamentului şi 90% din răspunsul maxim este atins după 2 săptămâni. Răspunsul maxim este atins de obicei după 4 săptămâni şi se menţine ulterior.
Eficacitate şi siguranţă clinică
Rosuvastatina este eficace la adulţii cu hipercolesterolemie, cu şi fără hipertrigliceridemie, indiferent de rasă, sex sau vârstă şi la anumite populaţii cum sunt diabeticii, sau pacienţii cu hipercolesterolemie familială.
Prin analiza datelor cumulate din studiile de fază III, rosuvastatina s-a demonstrat a fi eficace la majoritatea pacienţilor cu hipercolesterolemie de tip IIa şi IIb (valoarea iniţială medie a LDL-C de aproximativ 4,8 mmol/l) atunci când tratamentul a urmărit atingerea valorilor ţintă recomandate de către Societatea Europeană de Ateroscleroză (EAS; 1998); aproximativ 80% din pacienţii trataţi cu 10 mg au atins nivelele ţintă recomandate de EAS pentru LDL-C (<3 mmol/l).
Într-un studiu mare care a inclus 435 pacienţi cu hipercolesterolemie familială - forma heterozigotă au fost trataţi cu rosuvastatina în doze între 20 mg şi 80 mg cu proiect de tatonare a dozelor. Toate dozele s-au demonstrat a avea efecte favorabile asupra parametrilor lipidici şi asupra valorilor ţintă terapeutice. După creşterea treptată până la o doză zilnică de 40 mg (la 12 săptămâni de tratament), concentraţia LDL-C s-a redus cu 53%. 33% din pacienţi au atins valorile recomandate pentru LDL-C de către EAS (<3 mmol/l).
Într-un studiu deschis, cu proiect de tatonare a dozelor, 42 pacienţi cu hipercolesterolemie familială - forma homozigotă au fost evaluaţi privind răspunsul lor la tratamentul cu doze de 20-40 mg rosuvastatina. În toată populaţia, scăderea medie a valorilor LDL-C a fost de 22%.
În studiile clinice cu un număr limitat de pacienţi, rosuvastatina a demonstrat o eficacitate suplimentară în ceea ce priveşte scăderea concentraţiei plasmatice a trigliceridelor atunci când a fost administrată în asociere cu fenofibrat şi în ceea ce priveşte creşterea concentraţiei plasmatice a HDLC atunci când a fost administrat în asociere cu niacina.
Într-un studiu clinic multi-centric, dublu-orb, placebo-controlat (METEOR) la 984 pacienţi cu vârsta cuprinsă între 45 şi 70 ani, cu risc scăzut de boală coronariană (definis ca risc Framingham <10% pe o perioadă de 10 ani), cu valori medii ale LDL-colesterolului de 4,0 mmol/l (154.5 mg/dl), dar cu ateroscleroză subclinică (detectată prin testul CIMT - Carotid Intima Media Thickness) au fost repartizaţi randomizat la tratament cu rosuvastatină 40 mg o dată pe zi sau placebo, timp de 2 ani. Rosuvastatina a încetinit semnificativ rata de evoluţie a grosimii intimei arterei carotidiene (CIMT) comparativ cu placebo cu -0,0145 mm/an [IÎ 95% -0.0196, -0.0093; p<0,0001]. Modificarea faţă de valorile bazale a fost de -0,0014 mm/an (-0.12%/an (fără semnificaţie statistică)) pentru rosuvastatină, comparativ cu evoluţia de +0,0131 mm/an (1,12%/an (p<0,0001)) pentru cei din grupul placebo. Nu a fost încă demonstrată relaţia directă între scăderea CIMT şi reducerea riscului evenimentelor cardiovasculare. Grupele de populaţie studiate în METEOR prezintă risc scăzut pentru boală coronariană şi nu reprezintă populaţia ţintă pentru doza de rosuvastatină de 40 mg. Această doză trebuie prescrisă numai la pacienţii cu hipercolesterolemie severă şi risc cardiovascular înalt.
Pentru justificarea utilizării statinelor în prevenţia primară, a fost efectuat un studiu de evaluare intervenţională cu rosuvastatină (JUPITER), asupra efectelor rosuvastatinei asupra incidenţei evenimentelor majore cardiovasculare aterosclerotice la 17802 bărbaţi (>50 ani) şi femei (>60 ani).
Participanţii la studiu au fost repartizaţi randomizat la tratament cu placebo (n=8901) sau rosuvastatină 20 mg o dată pe zi (n=8901) şi au fost urmăriţi, în medie, timp de 2 ani.
Concentraţia de LDL-colesterol a fost redusă cu 45% (p<0,001) în grupul tratat cu rosuvastatină, comparativ cu grupul tratat cu placebo.
Într-o analiză post-hoc a unor sub-grupe de pacienţi cu risc crescut cu scor de risc Framingham de bază de >20% (1558 subiecţi), s-a observat o reducere semnificativă a obiectivului combinat de mortalitate cardiovasculară, accident vascular cerebral şi infarct de miocard (p=0,028) la tratamentul cu rosuvastatină versus placebo. Reducerea absolută a ratei riscului de eveniment per 1000 pacient-ani a fost de 8,8. În cadrul acestui grup de risc înalt, mortalitatea totală a rămas nemodificată (p=0,193). Într-o analiză post-hoc a unor sub-grupe de pacienţi cu risc crescut (9302 subiecţi) cu un scor de risc de bază de 5% (extrapolat pentru a include subiecţi cu vârsta de peste 65 ani), s-a observat o reducere semnificativă a obiectivului combinat final de mortalitate cardiovasculară, accident vascular cerebral şi infarct de miocard (p=0,0003) la grupul tratat cu rosuvastatină, faţă de cel tratat cu placebo. Reducerea absolută a ratei riscului de eveniment per 1000 pacient-ani a fost de 5,1. În cadrul acestui grup de risc înalt, mortalitatea totală a rămas nemodificată (p=0,076).
În studiul JUPITER, 6,6% dintre pacienţii trataţi cu rosuvastatină şi 6,2% dintre subiecţii trataţi cu placebo au întrerupt studiul din cauza unui eveniment advers. Cele mai frecvente reacţii adverse care au determinat întreruperea tratamentului au fost: mialgie (0,3% la rosuvastatină, 0,2% la placebo), durere abdominală (0,03% la rosuvastatină, 0,02% la placebo) şi erupţie cutanată tranzitorie (0,02% la rosuvastatină, 0,03% la placebo). Cele mai frecvente reacţii adverse cu proporţie mai mare sau egală cu grupul placebo au fost: infecţie urinară (8,7% la rosuvastatină, 8,6% la placebo), nazofaringită (7,6% la rosuvastatină, 7,2% la placebo), durere dorsală (7,6% la rosuvastatină, 6,9% la placebo) şi mialgie (7,6% la rosuvastatină, 6,6% la placebo).
Copii şi adolescenţi
Într-un studiu dublu-orb, randomizat, multi-centric, placebo-controlat, de 12 săptămâni (n=176,97 de sex masculin şi 79 de sex feminin) urmat de o fază deschisa de încă 40 săptămâni (n=173,96 de sex masculin şi 77 de sex feminin), au participat pacienţi cu vârsta de 10 până la 17 ani (în stadiu Tanner II-V, iar fetele la cel puţin 1 an după menarhă) cu hipercolesterolemie familială heterozigotă, care au primit rosuvastatină 5, 10 sau 20 mg pe zi sau placebo, timp de 12 săptămâni, după care toţi au primit rosuvastatină o dată pe zi, timp de 40 săptămâni. La intrarea în studiu, aproximativ 30% dintre pacienţi aveau vârsta între 10>13 ani şi aproximativ 17%, 18%, 40%, şi 25% au fost în stadii Tanner II, III, IV, şi V.
Concentraţia LDL-colesterolului a fost redusă cu 38,3%, 44,6% şi 50,0% în urma tratamentului cu rosuvastatină în doză de 5, 10 şi, respectiv, 20 mg, comparativ cu 0,7% pentru cei din grupul placebo.
La sfârşitul celor 40 săptămâni de fază deschisă, cu creşterea dozei de până la maxim 20 mg o dată pe zi, 70 din cei 173 pacienţi (40,5%) au atins nivelul ţintă al concentraţiei LDL-colesterolului de sub 2,8 mmol/l.
După cele 52 săptămâni de tratament ale studiului, nu au fost observate efecte asupra creşterii, greutăţii corporale, indexului masei corporale sau a maturizării sexuale (vezi pct. 4.4). Datele din studii clinice la copii şi adolescenţi sunt limitate, iar efectele pe termen lung ale rosuvastatinei (>1 an) asupra pubertăţii sunt necunoscute. Acest studiu (n=176) nu a fost conceput pentru comparaţii ale reacţiilor adverse rare.
Proprietăţi farmacocinetice
Absorbţie
Concentraţiile plasmatice maxime de rosuvastatină se ating după 5 ore de la administrarea orală. Biodisponibilitatea absolută este de aproximativ 20%.
Distribuţie
Rosuvastatina este captată extensiv de ficat, care este principalul loc de sinteză a colesterolului şi al clearance-ului LDL-C. Volumul aparent de distribuţie al rosuvastatinei este de aproximativ 134 l. Aproximativ 90% din rosuvastatină se leagă de proteinele plasmatice, în principal de albumină.
Metabolizare
Rosuvastatina este metabolizată limitat (de aproximativ 10%). Studiile in vitro asupra metabolismului, în care s-au folosit hepatocite umane au arătat că rosuvastatina este un substrat slab pentru metabolizarea dependentă de citocromul P450. Principala izoenzimă implicată a fost CYP2C9 iar izoenzimă 2C19, 3A4 şi 2D6 au fost implicate într-o măsură mai mică. Principalii metaboliţi identificaţi sunt N-desmetil- şi lacton-metaboliţii. Metabolitul N-desmetil este cu aproximativ 50% mai puţin activ decât rosuvastatina, în timp ce metabolitul lactonă este considerat inactiv clinic.Rosuvastatina este responsabilă de inhibarea activităţii a mai mult de 90% din HMG-CoA reductaza circulantă.
Eliminare
Aproximativ 90% din doza de rosuvastatină se excretă nemodificată în materiile fecale (constând în substanţă activă absorbită şi neabsorbită), iar restul este excretată prin urină. Aproximativ 5% este excretată nemodificat prin urină. Timpul de înjumătăţire plasmatică prin eliminare este de aproximativ 20 ore. Timpul de înjumătăţire plasmatică prin eliminare nu creşte la administrarea unor doze mai mari. Media geometrică a clearance-ului plasmatic este de aproximativ 50 litri/oră (coeficient de variaţie 21,7%). Ca şi în cazul altor inhibitori de HMG-CoA reductază, captarea hepatică a rosuvastatinei implică transportorul membranar OATP-C. Acest sistem de transport este important pentru eliminarea hepatică a rosuvastatinei.
Liniaritate/nonliniaritate
Expunerea sistemică la rosuvastatină creşte proporţional cu doza administrată. Administrarea mai multor doze zilnice nu este urmată de modificări ale parametrilor farmacocinetici.
Grupe speciale de populaţie
Vârstă şi sex
Nu s-a constatat nici o modificare semnificativă clinic, legată de vârstă sau de sex, în ceea ce priveşte farmacocinetica rosuvastatinei la adulţi. Farmacocinetica rosuvastatinei la copii şi adolescenţi cu hipercolesterolemie familială heterozigotă a fost similară celei a voluntarilor tineri.
Rasă
Studii de farmacocinetică au aratat o creştere de aproximativ 2 ori a valorilor medii ale ASC şi Cmax la pacienţii asiatici (Japonia, China, Filipine, Vietnam şi Coreea) comparativ cu pacienţii de rasa albă; populaţiile asiatice-indiene prezintă o creştere de aproximativ 1,3 ori a valorilor medii ale ASC şi Cmax. O analiză populaţională de farmacocinetică la pacienţii de rasă albă şi neagră nu a arătat diferenţe clinic relevante ale parametrilor farmacocinetici.
Insuficienţă renală
Într-un studiu efectuat la pacienţi în diferite grade ale insuficienţei renale, insuficienţa renală uşoară până la moderată nu influenţează concentraţia plasmatică a rosuvastatinei sau a metabolitului său Ndesmetil. Pacienţii cu insuficienţă renală severă (clearance-ul la creatinină < 30 ml/min) au prezentat o concentraţie plasmatică de rosuvastatină de 3 ori mai mare şi o concentraţie plasmatică a metabolitului N-desmetil de 9 ori mai mare, comparativ cu voluntarii sănătoşi. Concentraţiile plasmatice ale rosuvastatinei, la starea de echilibru, la pacienţii care efectuează hemodializă au fost cu aproximativ 50% mai mari comparativ cu voluntarii sănătoşi.
Insuficienţă hepatică
Într-un studiu efectuat la pacienţi cu diferite grade de afectare hepatică, nu s-a evidenţiat creşterea expunerii la rosuvastatină la pacienţii cu un scor Child-Pugh de 7 sau mai mic. Cu toate acestea, 2 pacienţi cu scoruri Child-Pugh de 8 şi 9 au prezentat o creştere a expunerii sistemice la rosuvastatină de cel puţin de 2 ori mai mare comparativ cu pacienţii care prezentau scoruri Child-Pugh mai mici. Nu au fost studiaţi pacienţi cu scoruri Child-Pugh mai mari de 9.
Copii şi adolescenţi
Parametrii farmacologici la copii şi adolescenţi cu hipercolesterolemie familială heterozigotă cu vârsta între 10 şi 17 ani nu au avut caracteristici deosebite. Un stdiu farmacocinetic de mici dimensiuni cu rosuvastatină (administrată sub formă de comprimate) la 18 copii a demonstrat că nivelul concentraţiei plasmatice la copii pare similar adulţilor. În plus, rezultatele arată că nu se aşteaptă modificări ale proporţionalităţii dozei.
Date preclinice de siguranţă
Datele preclinice din studiile convenţionale de siguranţă farmacologică,de toxicitate după doze repetate, de genotoxicitate şi potenţial carcinogen nu au arătat nici un risc deosebit pentru om. Nu au fost evaluate testele specifice asupra hERG. Reacţiile adverse care nu au apărut în studiile clinice, însă au fost prezente la animale la administrarea unor doze similare celor recomandate au fost următoarele: în studiile de toxicitate la doze repetate, s-au produs modificări histopatologice hepatice probabil datorate acţiunii farmacologice a rosuvastatinei, la şoarece, şobolan şi, în mai mică măsură, la nivelul veziculei biliare la câini, dar nu şi la maimuţe. În plus, la maimuţe şi câini, la doze crescute, a fost observată toxicitate testiculară. Într-un studiu efectuat la şobolan, pre- şi postnatal, toxicitatea asupra funcţiei de reproducere a fost evidentă, ţinând cont de dimensiunile, greutatea feţilor şi supravieţuirea puilor reduse. Aceste efecte au fost observate la doze toxice pentru mame la o expunere sistemică de câteva ori mai mare decât nivelul terapeutic de expunere sistemică.
PROPRIETĂŢI FARMACEUTICE
Lista excipienţilor
Nucleu
Lactoză anhidră
Celuloză microcristalină
Crospovidonă
Stearat de magneziu
Dioxid de siliciu coloidal anhidru.
Film
Lactoză monohidrat
Dioxid de titan (E171)
Macrogol 6000
Copolimer butyl-metacrilat
Incompatibilităţi
Nu este cazul.
Perioada de valabilitate
3 ani.
Precauţii speciale pentru păstrare
Acest medicament nu necesită precauţii speciale de păstrare.
A se păstra în ambalajul original, pentru a fi protejat de lumină.
Natura şi conţinutul ambalajului
Blistere perforate din OPA-Al-PVC-Al): cutie cu 10, 14, 20, 28, 30, 56, 60, 84, 90, 98 şi 100 comprimate filmate.
Este posibil ca nu toate mărimile de ambalaj să fie comercializate.
DEŢINĂTORUL AUTORIZAŢIEI DE PUNERE PE PIAŢĂ
KRKA, d.d., Novo mesto, Šmarješka cesta 6, 8501 Novo mesto, Slovenia
Livrare maxim în 24 ore în Chișinău!

În Chișinău
Gratuit sau 50 MDL pentru comenzi mai mici de 450 MDL

În restul Moldovei
Gratuit sau 50 MDL pentru comenzi mai mici de 450 MDL

În orice farmacie
Gratuit pentru orice comandă, indiferent de sumă
Informații importante despre produs
S.R.L. Farmacia Familiei își rezervă dreptul de a modifica în orice moment, fără notificare prealabilă, descrierile și caracteristicile produselor.
Imaginile și informațiile prezentate pe site au caracter informativ și orientativ și pot conține erori sau neconcordanțe. Depunem eforturi pentru asigurarea acurateței acestora, însă nu garantăm lipsa erorilor.
Înainte de utilizarea sau consumul oricărui produs, se recomandă consultarea medicului sau farmacistului.
Disponibil în următoarele locații Disponibilitate
244 farmaciiLuni - Duminica, 08:00 - 20:00
Marți - Duminică, 07:30 - 16:00
Luni, 08:00-14:00
Luni - Vineri, 08:00 - 18:00
Sâmbătă, 08:00 - 13:00
Luni - Vineri, 08:00 - 19:00
Sâmbătă - Duminica, 08:00 - 16:00
Luni - Vineri, 08:00 - 17:00
Sîmbătă, 08:00 - 13:00
Produse similare



Ce spun clienții
Lasă recenzia ta

KRKA d.d., Novo mesto
Roswera 20mg comp.film.
Împărtășiți-vă experiența cu acest produs
Vezi toate review-urile
Din aceiași serie


Cele mai vândute